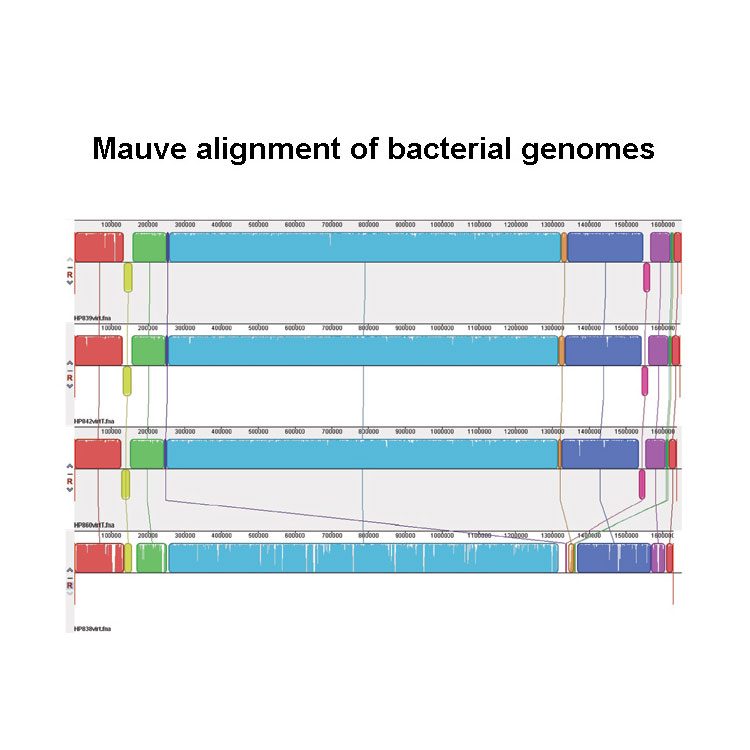
Evaluating assembly and variant calling software for strain-resolved analysis of large DNA viruses. Deng Z-L, Dhingra A, Fritz A, Götting J, Münch PC, Steinbrück L, Schulz TF, Ganzenmüller T, McHardy AC. Brief Bioinform 2021 ; 22; 3
Human Cytomegalovirus Genome Diversity in Longitudinally Collected Breast Milk Samples. Götting J ,Lazar K ,Suárez NM, Steinbrück L, Rabe T, Goelz R ,SchulzTF ,Davison AJ, Hamprecht K, Ganzenmueller T. Front Cell Infect Microbiol 2021; 11
Human cytomegalovirus multiple-strain infections and viral population diversity in haematopoietic stem cell transplant recipients analysed by high-throughput sequencing. Dhingra A, Götting J, Varanasi PR, Steinbrueck L, Camiolo S, Zischke J, Heim A, Schulz TF, Weissinger EM, Kay-Fedorov PC, Davison AJ, Suárez NM, Ganzenmueller T. Med Microbiol Immunol 2021 ; 210; 44717
Why? – Successful Pseudomonas aeruginosa clones with a focus on clone C. Lee C, Klockgether J, Fischer S, Trcek J, Tümmler B, Römling U. FEMS Microbiology Reviews 2020 November, Volume 44, Issue 6, Pages 740–762
Intraclonal competitive fitness of longitudinal cystic fibrosis Pseudomonas aeruginosa airway isolates in liquid cultures. Cramer N, Fischer S, Hedtfeld S, Dorda M, Tümmler B. Environmental Microbiology 2020 Jan 26
Emerging therapies against infections with Pseudomonas aeruginosa. Tümmler B. F1000Res. 2019 Aug 7; 8: F1000 Faculty Rev-1371.
Novel Virus Related to Kaposi’s Sarcoma–Associated Herpesvirus from Colobus Monkey. Dhingra A, Ganzenmueller T, Hage E, Suárez NM, Mätz-Rensing K, Widmer D, Pöhlmann S, Davison AJ, Schulz TF, Kaul A. Emerg Infect Dis. 2019 Aug; 25(8): 1548–1551.
Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. Suárez NM , Wilkie GS , Hage E , Camiolo S , Holton M , Hughes J , Maabar M , Vattipally SB , Dhingra A , Gompels UA, Wilkinson GWG , Baldanti F , Furione M , Lilleri D , Arossa A , Ganzenmueller T , Gerna G , Hubáček P , Schulz TF, Wolf D, Zavattoni M, DavisonAJ. J Infect Dis. 2019 Jul 31;220(5):781-791.
Within-host evolution of Helicobacter pylori shaped by niche-specific adaptation, intragastric migrations and selective sweeps. Ailloud F, Didelot X, Woltemate S, Pfaffinger G, Overmann J, Bader R C, Schulz C, Malfertheiner P, Suerbaum S. Nat Commun. 2019 May 22;10(1):2273.
A unique methylation pattern by a type I HsdM methyltransferase prepares for DpnI rare cutting sites in the Pseudomonas aeruginosa PAO1 genome. Fischer S, Römling U, Tümmler B. FEMS Microbiology Letters 2019 March, Volume 366, Issue 5, fnz053
The core genome m5C methyltransferase JHP1050 (M.Hpy99III) plays an important role in orchestrating gene expression in Helicobacter pylori. Estibariz I, Overmann A, Ailloud F, Krebes J, Josenhans C, Suerbaum S. Nucleic Acids Res. 2019 Mar 18;47(5):2336-2348
Long-term Microevolution of Pseudomonas aeruginosa Differs Between Mildly and Severely Affected Cystic Fibrosis Lungs. KlockgetherJ, Cramer N, Fischer S, Wiehlmann L, Tümmler B. Am J Respir Cell Mol Biol. 2018 Aug;59(2):246-256
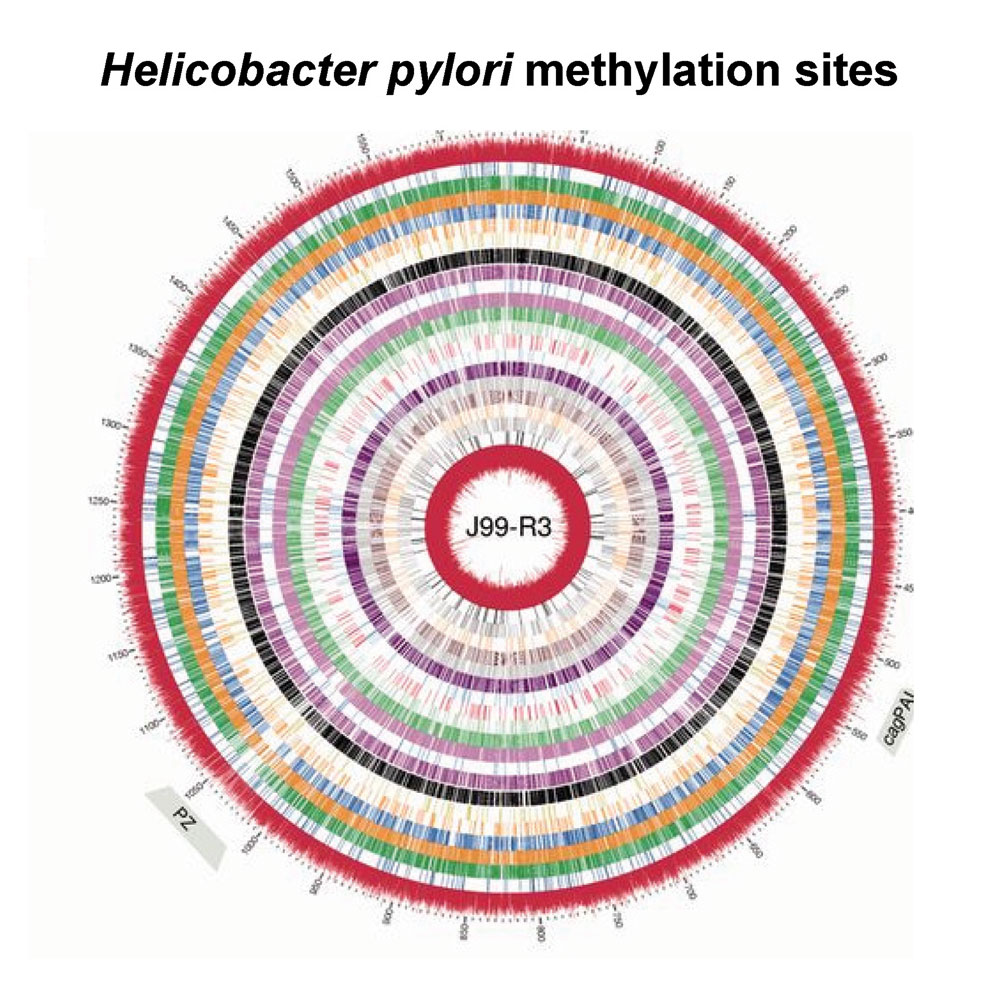
Genome and Methylome Variation in Helicobacter pylori With a cag Pathogenicity Island During Early Stages of Human Infection. Nell S, Estibariz I, Krebes J, Bunk B, Graham DY, Overmann J, Song Y, Spröer C, Yang I, Wex T, Korlach J, Malfertheiner P, Suerbaum S. Gastroenterology. 2018 Feb;154(3):612-623.e7.
Characterization of human cytomegalovirus genome diversity in immunocompromised hosts by whole genomic sequencing directly from clinical specimens. Hage E, Wilkie GS, Linnenweber-Held S, Dhingra A, Suárez NM, Schmidt JJ, Kay-Fedorov P, Mischak-Weissinger E, Heim A, Schwarz A, Schulz TF, Davison AJ, Ganzenmueller T. J Infect Dis. 2017 Mar 27
Filtration and Normalization of Sequencing Read Data in Whole-Metagenome Shotgun Samples. Chouvarine P, Wiehlmann L, Moran Losada P, DeLuca DS, Tümmler B. PLoS ONE 2016 11(10): e0165015.
Expanding the Host Range of Hepatitis C Virus through Viral Adaptation. von Schaewen M, Dorner M, Hueging K, Foquet L, Gerges S, Hrebikova G, Heller B, Bitzegeio J, Doerrbecker J, Horwitz JA, Gerold G, Suerbaum S, Rice CM, Meuleman P, Pietschmann T, Ploss A. MBio. 2016 Nov 8;7(6). pii: e01915-16.
SNP synteny analysis of Staphylococcus aureus and Pseudomonas aeruginosa population genomics. Losada PM, Tümmler B. FEMS Microbiol Lett. 2016 Oct 3. pii: fnw229
Three-base periodicity of sites of sequence variation in Pseudomonas aeruginosa and Staphylococcus aureus core genomes. Morán Losada P, Fischer S, Chouvarine P, Tümmler B. FEBS Lett. 2016 Oct;590(20):3538-3543.
Genome-wide analysis of chromosomal import patterns after natural transformation of Helicobacter pylori. Bubendorfer S, Krebes J, Yang I, Hage E, Schulz TF, Bahlawane C, Didelot X, Suerbaum S. Nat Commun. 2016 Jun 22;7:11995.
Different gastric microbiota compositions in two human populations with high and low gastric cancer risk in Colombia. Yang I, Woltemate S, Piazuelo MB, Bravo LE, Yepez MC, Romero-Gallo J, Delgado AG, Wilson KT, Peek RM, Correa P, Josenhans C, Fox JG, Suerbaum S. Sci Rep. 2016 Jan 5;6:18594.
Intraclonal genome diversity of the major Pseudomonas aeruginosa clones C and PA14. Fischer S, Klockgether J, Losada PM, Chouvarine P, Cramer N, Davenport CF, Dethlefsen S, Dorda M, Goesmann A, Hilker R, Mielke S, Schönfelder T, Suerbaum S, Türk O, Woltemate S, Wiehlmann L, Tümmler B. Environ Microbiol Rep. 2015 Dec 28.
Foxp3+ T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation.. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, Föhse L, Prinz I, Pezoldt J, Suerbaum S, Sparwasser T, Hamann A, Floess S, Huehn J, Lochner M. Mucosal Immunol. 2015 Aug 26
Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection… Hernández PP, Mahlakõiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, Gronke K, Ryffel B, Hölscher C, Dumoutier L, Renauld JC, Suerbaum S, Staeheli P, Diefenbach A. Nat Immunol. 2015 Jul;16(7):698-707.
A clonotypic Vγ4Jγ1/Vδ5Dδ2Jδ1 innate γδ T-cell population restricted to the CCR6⁺CD27⁻ subset. Kashani E, Föhse L, Raha S, Sandrock I, Oberdörfer L, Koenecke C, Suerbaum S, Weiss S, Prinz I. Nat Commun. 2015 Mar 13;6:6477.
Intestinal mucus affinity and biological activity of an orally administered antibacterial and anti-inflammatory peptide.. Dupont A, Kaconis Y, Yang I, Albers T, Woltemate S, Heinbockel L, Andersson M, Suerbaum S, Brandenburg K, Hornef MW. Gut. 2015 Feb;64(2):222-32.
A human adenovirus species B subtype 21a associated with severe pneumonia. Hage E, Huzly D, Ganzenmueller T, Beck R, Schulz TF, Heim A. J Infect. 2014 Nov;69(5):490-9.
TRIF signaling drives homeostatic intestinal epithelial antimicrobial peptide expression. Stockinger S, Duerr CU, Fulde M, Dolowschiak T, Pott J, Yang I, Eibach D, Bäckhed F, Akira S, Suerbaum S, Brugman M, Hornef MW. J Immunol. 2014 Oct 15;193(8):4223-34.
The complex methylome of the human gastric pathogen Helicobacter pylori. Krebes J, Morgan RD, Bunk B, Spröer C, Luong K, Parusel R, Anton BP, König C, Josenhans C, Overmann J, Roberts RJ, Korlach J, Suerbaum S. Nucleic Acids Res. 2014 Feb;42(4):2415-32. doi: 10.1093/nar/gkt1201. Epub 2013 Dec 2.
Role of energy sensor TlpD of Helicobacter pylori in gerbil colonization and genome analyses after adaptation in the gerbil. Behrens W, Schweinitzer T, Bal J, Dorsch M, Bleich A, Kops F, Brenneke B, Didelot X, Suerbaum S, Josenhans C. Infect Immun. 2013 Oct;81(10):3534-51. doi: 10.1128/IAI.00750-13. Epub 2013 Jul 8.
Evaluating a ligation-mediated PCR and pyrosequencing method for the detection of clonal contribution in polyclonal retrovirally transduced samples. Brugman MH, Suerth JD, Rothe M, Suerbaum S, Schambach A, Modlich U, Kustikova O, Baum C. Hum Gene Ther Methods. 2013 Apr;24(2):68-79. doi: 10.1089/hgtb.2012.175. Epub 2013 Mar 14.
Advances in computational analysis of metagenome sequences. Davenport CF, Tümmler B. Environ Microbiol. 2013 Jan;15(1):1-5. doi: 10.1111/j.1462-2920.2012.02843.x. Epub 2012 Aug 8.
Genometa–a fast and accurate classifier for short metagenomic shotgun reads. Davenport CF, Neugebauer J, Beckmann N, Friedrich B, Kameri B, Kokott S, Paetow M, Siekmann B, Wieding-Drewes M, Wienhöfer M, Wolf S, Tümmler B, Ahlers V, Sprengel F. PLoS One. 2012;7(8):e41224. doi: 10.1371/journal.pone.0041224. Epub 2012 Aug 21.
Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. Lindner C, Wahl B, Föhse L, Suerbaum S, Macpherson AJ, Prinz I, Pabst O. J Exp Med. 2012 Feb 13;209(2):365-77. doi: 10.1084/jem.20111980. Epub 2012 Jan 16.
Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, Wang TC, Fox JG. Gastroenterology. 2011 Jan;140(1):210-20. doi: 10.1053/j.gastro.2010.09.048. Epub 2010 Oct 13.